

")
Publication History
Submitted: April 04, 2023
Accepted: April 20, 2023
Published: May 01, 2023
Identification
D-0114
Citation
Pradip Rijal, Aatiqa Tariq & Syeda Hajra Batool (2023). The Study of Differential Expression of Genes Controlling Reproductive Function in Immune Cells of PCOS Women. Dinkum Journal of Medical Innovations, 2(05):157-169.
Copyright
© 2023 DJMI. All rights reserved
157-169
The Study of Differential Expression of Genes Controlling Reproductive Function in Immune Cells of PCOS WomenOriginal Article
Pradip Rijal 1*, Aatiqa Tariq 2, Syeda Hajra Batool 3
- Sunsari Technical College, Nepal; rijal.pradipn@gmail.com
- Hazara University, Mansehra, Pakistan; aatiqa.tariq@yahoo.com
- Lahore Garrison University, Pakistan; meetwithsyeda@gmail.com
* Correspondence: rijal.pradipn@gmail.com
Abstract: It is believed that immune responses are a major factor in the pathophysiology of PCOS, or polycystic ovarian syndrome. However, nothing is known about the properties of T lymphocyte subsets in PCOS. Here, we used RNA sequencing analysis to examine the gene’s function in PCOS pathogenesis as well as the differential expression of T-Helper cell genes. Much like the introduction of NGS technologies has revolutionised DNA sequencing, RNA-Seq approaches have allowed us to study the transcriptome of cells and tissues with unprecedented specificity and sensitivity. Our research led us to the conclusion that the pathogenesis of PCOS is associated with differentially expressed genes, indicating that immune cells play a major role in modulating the disease state through a variety of molecular mechanisms.
Keywords: Polycystic Ovary Syndrome (PCOS), reproductive functions, gene controlling, immune cells, women
- INTRODUCTION
PCOS is a long-term medical illness that affects various bodily functions, including reproduction, by altering the body’s overall metabolism. Furthermore, it is a hormonal condition that is characterised by polycystic ovary, hyperandrogenemia [2], and irregular ovulation. It is thought to be the primary cause of female infertility [1]. Moreover, a number of other variables, including as insulin resistance (IR) [3], type 2 diabetes mellitus (T2DM) [4], cardiovascular disease [5], and endometrial cancer [6], are thought to be linked to PCOS. Its management has been the subject of a wealth of research in recent years, spanning from the psychosocial consequences of illnesses to the treatment of ovulatory infertility and reproductive impacts [7]. The immune system serves as a defence mechanism that shields the body against disease by containing a variety of biological structures. A weakened immune system has been linked to a number of diseases, including chronic low-grade inflammation, which has been linked to PCOS14,15. It also includes increased leukocyte counts, endothelial dysfunction, and abnormal pro-inflammatory cytokines like interferon-gamma [IFN-γ] and interleukin-2 [IL-2]. Recent research has demonstrated that a number of cytokines, such as IFN-γ, TNF-α, IL-2, IL-4, IL-5, and IL-10, have a markedly detrimental effect on fertility, indicating a possible connection between immune system function and female infertility [6]. The immune systems control polycystic ovarian syndrome, according to recent studies [8]. Significantly, it was discovered that human pre-ovulatory cells contained large numbers of immune-competent cells, such as dendritic cells, DCs, T cells, B cells, neutrophils, and macrophages [9–12]. Furthermore, there is emerging recognition that immunological dysregulation contributes to PCOS16. The main component of lymphocytes, T cells, perform several biological functions, the most important of which are related to the body’s immune response. Additionally, they can increase the duration of the immune response by either directly eliminating the target cells or by releasing lymphatic factors [13, 14]. The primary modulators of immunological responses, including pro- and anti-inflammatory activities, are CD4+ Th cells. The distinct costimulatory cues and cytokines affect activated CD4+ T cells’ ability to develop into Th cells [15]. It is possible for IL-12 to direct the division of T cells into Th1 cells [16]. Similarly, Th2 cells and T cells, which mediate immunological tolerance, differ from one another due to the coordinated actions of IL-4 and IL-13 [17]. Estradiol and androgens in the bloodstream are strongly linked to inflammation [18], as evidenced by the discovery that PCOS patients’ follicular fluid had far lower amounts of IL-13 than did women with regular ovulation. Nonetheless, there was a notable rise in IL-12 concentrations [19], which could result in a transition from Th2 to Th1 cells [20, 21]. Furthermore, it has been demonstrated that progesterone spike lowers the levels of inflammatory cytokines in the luteal phase, but oestrogen increases the release of these molecules in Th1 cells, including TNF α, IL-6, and interferon-gamma (IFNγ). Because numerous follicles aggregate in PCOS patients, there is a significant degree of oestrogen without progesterone resistance. Th0 cells can differentiate into Th17 cells more easily when STAT3 is activated and RORα and RORγ expression is increased [22, 23]. This is because these proteins are major transcription factors of Th17 cells. While some research suggests that Th17 associated with PCOS declines in animal models [24, 25], other findings highlight the expansion of proinflammatory Th17 subtypes in both blood and kidneys [25]. On the other hand, the cause of human PCOS is unclear. In this circumstance, IL-6 can efficiently drive angiogenesis, which promotes blood vessel construction and raises follicle-stimulating hormone concentrations, while also inhibiting the generation of TNF α [26]. Technologies for high-throughput sequencing find extensive use in the field of biology [27]. Through the methodical application of transcriptomes, genomes, and epigenomes, these technologies generate millions of short reads [28]. For example, RNA-Seq has been proven to be essential for cataloguing the variety of novel transcript species, such as long non-coding RNA, miRNA, siRNA, and other tiny classes of RNA [30] and for enabling the determination of innovative transcription families [29]. Most RNA-Seq experiments on a high throughput platform start with an amount of pure RNA, compress it, transform it to cDNA, and subsequently sequencing it [29]. Our goal is to perform RNASeq analysis to look into the diverse ways that T-Helper cell genes are expressed and to determine how these differences connect to the pathophysiology of PCOS.
- MATERIALS AND METHODS
RNA-sequencing was used to investigate the transcriptome and genomic DNA methylation in T helper cells [31]. A detailed background on the examination of particular genome-wide DNA methylation and the methylated CpG sites involved in pathways associated to immune cell and reproductive function is provided by the dataset submitted to the ENA Database and the SRA Database in the NCBI. We are now going to make a small modification to the previous study. The objective of our research is to do RNASeq analysis to examine the variations in T-Helper cell gene expression and evaluate the function of these genes in the pathophysiology of PCOS. The majority of RNA-Seq studies involve the following essential steps in their data analytics. Analysing in a similar manner, we discovered the following findings. Library preparation and sequencing itself may result poor-quality data[32]. PCR artifacts[32], untrimmed adapter sequences[33], sequence-specific bias, and other possible contaminants can also lead to poor data quality. The presence of technical or low quality sequences can affect downstream analysis and data interpretation, leading to erroneous results [34]. To assess the quality of raw data, a number of tools have been created, including PRINSEQ [35] and FastQC [36].
- RESULTS
The goal of FastQC is to make it simple to conduct quality control tests on raw sequence data. It provides a modular set of analyses that may be used to quickly determine whether raw sequencing data has any problems that should be known before conducting additional study.
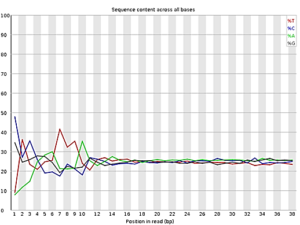
Figure 1: FastQC result for quality check
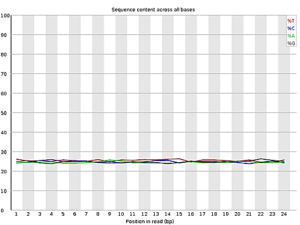
Figure 2: FastQC results after trimming the head and tail regions
After the accuracy of the data has been confirmed, reads that contain low-quality bases, including adapter sequences, and other contaminating sequences should be processed to eliminate them. For these kinds of work, two examples of tools that can be utilised are Cut adapt [37] and Trimmomatic [38]. After completing the previously described steps, the sequencing data is ready for a subsequent assessment. We ran FastQC on each paired end dataset utilised in our study, and the results included a range of quality assessments and basic read statistics. Most of the sequences earned a score that was acceptable for their quality. Nevertheless, we used Trimmomatic to tidy up the GC content and its distribution. Reports created from both the raw and quality-filtered data files included pertinent information such as adaptor type, numbers, sequence length transportation, GC content, Phred (quality) scores, and sequence duplication levels. To ascertain the actual origins of short sequence reads produced by sequencers with regard to the reference, they must be compared to a reference genome. A plethora of algorithms, including TopHat2 [39], STAR [40], [41], GSNAP [42], OSA [43], and Map Splice [44], have been created recently for mapping reads. In our investigation, we performed mapping by RNA star using the UCSC GRCh38/Hg38 reference genome. Introduced in 2001[45], the UCSC Genome Browser website (http://genome.ucsc.edu) and related tools exhibit the human genome assembly process. Since then, they have expanded to provide access to sophisticated genome assemblies and annotations for researchers and scholars around the globe. The most regularly utilized research function was read alignment to the reference genome using the simple RNA Star technique. These alignments serve as the foundation for a plethora of subsequent investigations, such as gene expression prediction and visualization. Finding the largest mappable region of reading is the main technique of the STAR alignment strategy [46]. The split/search/extend function, which is powered by the sequencing quality scores, is another essential part of the process. It makes it possible to accurately align reads that have one or more splice junctions or a significant number of sequencing errors. The presence of repetitive portions, assembly errors, and assembly gaps in a reference genome may make it difficult for a fraction of reads to achieve this [47]. Moreover, RNA-Seq libraries made from transcribed RNA and exon-exon spanning reads do not contain intronic sequences. It reads the “exon-exon” junctions to divide over hundreds of intronic sequence bases while aligning the sequences to a reference genome. The advantages of mapping RNA-Seq reads using a reference genome have been well-illustrated in previous research [48]–[50]. Furthermore, a comprehensive evaluation of the mapping of RNA-Seq reads and gene quantifications using Ensemble [53], UCSC recognized Genes [52], and Ref-Seq Gene[51] has been reported. Numerous methods for gene inference have been developed as RNA-Seq has grown in popularity in molecular biology labs. These methods include Feature Counts (57), Ht-seq (58), Cufflinks (55), IsoEM (56), RSEM (54), and Cufflinks (55). When it comes to counting, the algorithms Feature Counts [57] and Ht-seq [58] perform similarly; however, feature-counts are far faster and need far less computer memory for gene-level summarization than Ht-seq. However, neither Feature Counts nor Ht-seq can attain transcript-level read counts because of the way they are implemented. We used a minimum alignment quality of 10 for Ht-seq, measuring the number of matched reads that spanned the exons of each gene using a BAM file and a GTF file. Following the measurement of the read counts, data normalisation is an essential stage in data processing. Careful consideration is required for this approach in order to guarantee accurate gene expression inference for further investigation. Since different samples typically have different sequencing depths or library sizes (the total amount of mapped reads)[59], it is not possible to directly compare the counts found across samples. Recalibration of the cumulative read counts is the most effective method to normalise the discrepancy in library sequencing sizes. But generally speaking, this kind of normalisation is inappropriate because RNA-Seq counts represent the gene abundances in a sample. The amount of reads mapped to a gene depends on the gene’s length and degree of expression, as well as the make-up of the RNA population being sampled. A small number of highly expressed genes can absorb a very substantial fraction of all the readings in a sample, repressing the counts of all the other genes. Because of this, the expression of these suppressed genes is typically lower than that of a sample with a more evenly distributed read distribution, which results in many genes that are incorrectly differentially expressed. Finding genes that are differently expressed between different sample classes is a basic research objective in many RNA-Seq studies. Recently, a number of algorithms have been developed expressly for the purpose of identifying differentially expressed genes (DEGs) from RNA-Seq findings. These methods include DESeq[60], edgeR[61], [62], GENE-Counter[63], NOISeq[64], NBPSeq[65], and Cuffdiff2[66]. In our work, we used DESeq2 and the data factor of Disease (PCOS) VS Control to normalise our datasets. We looked at the MA-Plot, p-values, and dispersion estimates. We also looked at the differences in expression for 20 top-ranked important genes on a Volcano plot. Twenty genes were discovered to be expressed differently in PCOS-affected and non-PCOS-affected women (Figure 3), with two upregulated and eighteen downregulated (Tables 1 & 2). It’s interesting to note that whereas proteins expressed by downregulated genes are not found in immune cells, those encoded by upregulated genes have little selectivity towards immune cells.
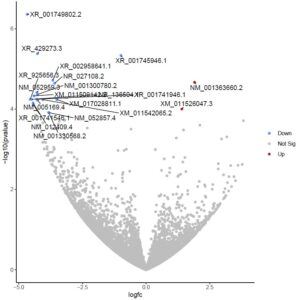
(A)
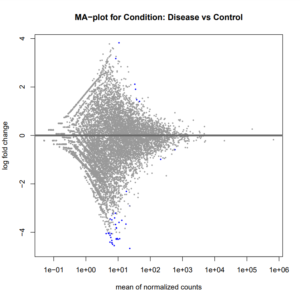
(B)
Figure 3: A) Volcano plots representing differentially expressed genes in T helper cells of women with PCOS in comparison to the women without PCOS. Red circles represent the upregulated genes while blue circles represent the downregulated genes. B) MA-plot representing Disease (PCOS) vs. Control condition.
TABLE 1: The list of up-regulated genes
| Symbol | Name | ENSEMBL ID | Chromosome |
| PSPC1 | Paraspeckle Component 1 | ENSG00000121390 | 13 |
| RNF125 | Ring Finger Protein 125 | ENSG00000101695 | 18 |
TABLE 2: The list of down regulated genes
| Symbol | Name | ENSEMBL ID | Chromosome |
| LINC02995 | Long intergenic non-protein coding RNA 2995 | ENSG00000253955 | 5 |
| PANX3 | Pannexin 3 | ENSG00000154143 | 11 |
| SEMA6A | Semaphorin | ENSG00000092421 | 5 |
| PHOX2A | Paired like homobox 2A
|
ENSG00000165462 | 11 |
| PRND | Prion like protein doppel | ENSG00000171864 | 20 |
| USB1 | U6 SnRNA biogenesis phosphodiesterase 1 | ENSG00000103005 | 16 |
| ZNF830 | Zinc Finger Protein 830 | ENSG00000198783 | 17 |
| TSHB | Thyroid stimulating hormone subunit beta | ENSG00000134200 | 1 |
Getting a list of DE genes is the first step towards learning more about the molecular mechanisms underlying disease or developmental phases, as well as biological insights into experimental procedures. Pathway enrichment research, for example, guarantees that the biological context of DE genes is understood. Furthermore, functional enrichment analyses are used by annotation databases including Gene Ontology (GO)[67], Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways[68], DAVID, and other commercial information structures like Ingenuity Pathway Analysis (IPA). We used the David Database to do functional annotations for our experiment. Ten items are enriched in molecular and cellular functions, whereas eight out of the twenty entries are enriched in biological processes. Based on functional annotation analysis, the majority of immune-related biological processes are found to have an enrichment of the elevated gene PSPC1. Similarly, RNF125, another elevated gene, has been discovered to be enriched in adaptive immunity. The ZNF830 gene is abundant in biological processes such as preantral ovarian follicle growth and ovarian follicle development, according to a study on gene ontology. Similar to this, the RNF125 gene controls the RIG-I signalling system, inhibits the generation of type I interferon, and aids in the adaptive immune response. Similarly, the innate immune response is activated by the PSPCI gene. Furthermore, it appears that PANX3 is involved in the upregulation control of the production of interleukin-1.
- DISCUSSION
It has been discovered that the overexpressed gene PSPC1 increases TGF-β production through its interaction with nuclear and phosphorylated Smad2/3, which in turn mediates TGF-β autocrine signaling[69]. TGF-β is aggressively deposited in the extracellular matrix as a sizable latent complex that awaits activation by integrins and proteases[70]. Following activation, it increases fibroblast activity, leading to enhanced collagen synthesis and deposition in stroma of both normal and fibrotic tissues[71], [72].The ovaries of women diagnosed with PCOS consistently exhibit all the signs of increased TGF-β activity, including increased thickness of the cortical and subcortical stroma and a greater number of ovarian capsules, or tunica albugineas[73]. The phenomena of local TGF-β dysregulation leading to reproductive disorders characterised by ovarian hyperandrogenism and anovulation is shown by the thicker and enlarged ovarian stroma. Furthermore, TGF-β pathways are essential for cell formation, particularly folliculogenesis, which is linked to PCOS. Furthermore, there is evidence connecting elevated TGF-β to the metabolic and cardiovascular disorders connected to PCOS [74]. As previously noted, an additional elevated gene, RNF125, may additionally activate the TGF-β1 SMAD3-1D1 signalling pathway, resulting in enhanced autocrine TGF-β1 signalling and a harmful effect in PCOS. Furthermore, RNF 125 has been linked to the stimulation of interleukin-36 receptor signalling [76], which is recognised to play a role in tissue fibrosis. Thorough investigation into these phenomena, which may be related to ovarian fibrosis, may help us understand its possible connection to PCOS. In terms of down-regulated genes, it is known that down-regulated long non-coding RNAs with elevated IL-18, which is connected to the clinical hallmark of PCOS—abrupt follicle growth and anovulation—contribute to insulin resistance in PCOS patients [77, 78]. Because oogenesis requires a high energy supply, the family of large pore-forming channels known as pannexins plays a crucial part in the process [79]. Specifically, it has been discovered that pannexin 3 regulates intracellular ATP/cAMP levels [80], which also tend to meet the need for an energy supply during oocyte growth. Therefore, dysregulation of the processes of oogenesis, folliculogenesis, oocyte maturation, and fertilisation can be linked to downregulation of pannexin 3, which may therefore result in PCOS-related infertility. The ZNF830 gene is found to be enriched in the biological processes of ovarian follicle development and preantral ovarian follicle growth through the gene ontology study. This finding explains why the downregulation of this specific gene has a significant correlation with the pathogenesis of PCOS, which ultimately results in infertility in women. The beta gene for the thyroid stimulating hormone subunit gives instructions on how to generate the beta subunit, which then binds with the alpha subunit to produce the thyroid hormone in its active state. Thus, down-regulating this gene has a detrimental influence on the thyroid stimulating hormone production, which in turn causes insufficient thyroid hormone release, which results in hypothyroidism. As a result, increased collagen deposition and polycystic-appearing ovaries are clinical signs of hypothyroidism [81]. Similarly, women with hypothyroidism have been reported to have noticeably larger ovarian volumes [82], as well as altered ovarian functioning and irregular menstruation [83].
- CONCLUSION
The development of RNA-Seq techniques has allowed us to examine the transcriptome of cells and tissues with unprecedented precision, in a manner similar to how the introduction of NGS technologies revolutionised our methods for sequencing DNA. However, the usual biases and mistakes present in the next-generation sequencing (NGS) methodology might also affect RNA-Seq research. Despite the fact that the genetics of PCOS are still poorly understood, computational analysis of these conditions can be examined in detail to remarkably identify the mechanisms underlying such syndromes through the research of differentially expressed genes.
REFERENCES
- “https://www.cdc.gov/reproductivehealth/infertility/.”
- Dadachanji, N. Shaikh, and S. Mukherjee, “Genetic Variants Associated with Hyperandrogenemia in PCOS Pathophysiology,” Genetics Research International, vol. 2018. Hindawi Limited, 2018. doi: 10.1155/2018/7624932.
- K. Song, Y. S. Hong, Y.-A. Sung, and H. Lee, “Insulin resistance according to β-cell function in women with polycystic ovary syndrome and normal glucose tolerance,” PLOS ONE, vol. 12, no. 5, pp. e0178120-, May 2017, [Online]. Available: https://doi.org/10.1371/journal.pone.0178120
- Y. Boudreaux, E. O. Talbott, K. E. Kip, M. M. Brooks, and S. F. Witchel, “Risk of T2DM and impaired fasting glucose among pcos subjects: Results of an 8-year follow-up,” Current Diabetes Reports, vol. 6, no. 1, pp. 77–83, 2006, doi: 10.1007/s11892-006-0056-1.
- Dokras, “Cardiovascular disease risk in women with PCOS,” Steroids, vol. 78, no. 8, pp. 773–776, 2013, doi: https://doi.org/10.1016/j.steroids.2013.04.009.
- Navaratnarajah, O. C. Pillay, and P. Hardiman, “Polycystic Ovary Syndrome and Endometrial Cancer,” Semin Reprod Med, vol. 26, no. 01, pp. 062–071, 2008.
- He et al., “Peripheral Blood Inflammatory-Immune Cells as a Predictor of Infertility in Women with Polycystic Ovary Syndrome,” J Inflamm Res, vol. 13, pp. 441–450, Aug. 2020, doi: 10.2147/JIR.S260770.
- Mobeen, N. Afzal, and M. Kashif, “Polycystic Ovary Syndrome May Be an Autoimmune Disorder,” Scientifica (Cairo), vol. 2016, p. 4071735, 2016, doi: 10.1155/2016/4071735.
- Wu et al., “Single-Cell Sequencing Reveals an Intrinsic Heterogeneity of the Preovulatory Follicular Microenvironment,” Biomolecules, vol. 12, no. 2, p. 231, Jan. 2022, doi: 10.3390/biom12020231.
- Maryam Majeed, Rubab Ali, Umm-e-Aimen & Aqsa Jabeen. Medical Staff, Workplace Bullying and Its Effects on the Performance. Dinkum Journal of Medical Innovations, 2(04):120-125.
- Marie Diack. Factors Influencing Relational Violence in Nepali Married Relationships. Dinkum Journal of Medical Innovations, 2(04):126-133.
- Gul Rukh Malik, Rubab Zahra & Tahir Rana. Autism Spectrum Disorders Frequency in Asian Countries. Dinkum Journal of Medical Innovations, 2(04):134-139.
- Parshu Ram Chaudhary, Asbin Bandhari & Parshu Kirby. Aeroallergens and Significant Environmental Pollutants: Aeroallergen Sensitivity Symptoms. Dinkum Journal of Medical Innovations, 2(04):140-149.
- Imran Mehfooz Khan, Aron Kumar & Pradeep Sahejpal. Extensive Management Medical Incidents Provided to Gallbladder Cancer Using Cholecystectomy Specimen. Dinkum Journal of Medical Innovations, 2(04):150-156.
- -H. Song et al., “Interleukin-12 receptor β2 from grass carp: Molecular characterization and its involvement in Aeromonas hydrophila-induced intestinal inflammation,” Fish & Shellfish Immunology, vol. 87, pp. 226–234, 2019, doi: https://doi.org/10.1016/j.fsi.2019.01.016.
- L. Lee et al., “Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization,” Sci Rep, vol. 9, no. 1, p. 3260, Mar. 2019, doi: 10.1038/s41598-019-40078-8.
- González, N. S. Rote, J. Minium, and J. P. Kirwan, “Increased Activation of Nuclear Factor κB Triggers Inflammation and Insulin Resistance in Polycystic Ovary Syndrome,” The Journal of Clinical Endocrinology & Metabolism, vol. 91, no. 4, pp. 1508–1512, Apr. 2006, doi: 10.1210/jc.2005-2327.
- Gallinelli, I. Ciaccio, L. Giannella, M. Salvatori, T. Marsella, and A. Volpe, “Correlations between concentrations of interleukin-12 and interleukin-13 and lymphocyte subsets in the follicular fluid of women with and without polycystic ovary syndrome,” Fertil Steril, vol. 79, no. 6, pp. 1365–1372, 2003, doi: 10.1016/s0015-0282(03)00344-3.
- O. Yang et al., “T Helper 17 Lineage Differentiation Is Programmed by Orphan Nuclear Receptors RORα and RORγ,” Immunity, vol. 28, no. 1, pp. 29–39, 2008, doi: https://doi.org/10.1016/j.immuni.2007.11.016.
- Gong et al., “Association between Th1/Th2 immune imbalance and obesity in women with or without polycystic ovary syndrome,” Gynecological Endocrinology, vol. 34, no. 8, pp. 709–714, Aug. 2018, doi: 10.1080/09513590.2018.1428301.
- D. Gamble et al., “Inhibition of polyamine synthesis and uptake reduces tumor progression and prolongs survival in mouse models of neuroblastoma.,” Sci Transl Med, vol. 11, no. 477, p. eaau1099, 2019, doi: 10.1126/scitranslmed.aau1099.
- Moulana, “Immunophenotypic profile of leukocytes in hyperandrogenemic female rat an animal model of polycystic ovary syndrome,” Life Sci, vol. 220, pp. 44–49, Mar. 2019, doi: 10.1016/j.lfs.2019.01.048.
- Mannerås et al., “A New Rat Model Exhibiting Both Ovarian and Metabolic Characteristics of Polycystic Ovary Syndrome,” Endocrinology, vol. 148, no. 8, pp. 3781–3791, Aug. 2007, doi: 10.1210/en.2007-0168.
- B. Ressler, B. E. Grayson, and R. J. Seeley, “Metabolic, behavioral, and reproductive effects of vertical sleeve gastrectomy in an obese rat model of polycystic ovary syndrome,” Obes Surg, vol. 24, no. 6, pp. 866–876, Jun. 2014, doi: 10.1007/s11695-013-1153-2.
- M. P. Daan et al., “Biomarker Profiles in Women with PCOS and PCOS Offspring; A Pilot Study,” PLoS One, vol. 11, no. 11, pp. e0165033–e0165033, Nov. 2016, doi: 10.1371/journal.pone.0165033.
- W. Soon, M. Hariharan, and M. P. Snyder, “High-throughput sequencing for biology and medicine,” Molecular Systems Biology, vol. 9. Blackwell Publishing Ltd, 2013. doi: 10.1038/msb.2012.61.
- Chari et al., “SIGMA2: A system for the integrative genomic multi-dimensional analysis of cancer genomes, epigenomes, and transcriptomes,” BMC Bioinformatics, vol. 9, Oct. 2008, doi: 10.1186/1471-2105-9-422.
- Hrdlickova, M. Toloue, and B. Tian, “RNA-Seq methods for transcriptome analysis,” Wiley Interdisciplinary Reviews: RNA, vol. 8, no. 1. Blackwell Publishing Ltd, Jan. 01, 2017. doi: 10.1002/wrna.1364.
- J. Hangauer, I. W. Vaughn, and M. T. McManus, “Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs,” PLoS Genet, vol. 9, no. 6, pp. e1003569–e1003569, Jun. 2013, doi: 10.1371/journal.pgen.1003569.
- Hiam et al., “Epigenetic Reprogramming of Immune Cells in Women With PCOS Impact Genes Controlling Reproductive Function,” The Journal of Clinical Endocrinology & Metabolism, vol. 104, no. 12, pp. 6155–6170, Dec. 2019, doi: 10.1210/jc.2019-01015.
- F. Hess et al., “Library preparation for next generation sequencing: A review of automation strategies,” Biotechnology Advances, vol. 41, p. 107537, 2020, doi: https://doi.org/10.1016/j.biotechadv.2020.107537.
- Schubert, S. Lindgreen, and L. Orlando, “AdapterRemoval v2: Rapid adapter trimming, identification, and read merging,” BMC Research Notes, vol. 9, no. 1, Feb. 2016, doi: 10.1186/s13104-016-1900-2.
- Sun et al., “Impact of library preparation on downstream analysis and interpretation of RNA-Seq data: comparison between Illumina PolyA and NuGEN Ovation protocol.,” PLoS One, vol. 8, no. 8, 2013, doi: 10.1371/journal.pone.0071745.
- Schmieder and R. Edwards, “Quality control and preprocessing of metagenomic datasets,” Bioinformatics, vol. 27, no. 6, pp. 863–864, Mar. 2011, doi: 10.1093/bioinformatics/btr026.
- “https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.”
- Martin, “CUTADAPT removes adapter sequences from high-throughput sequencing reads,” EMBnet J, vol. 17, Aug. 2011, doi: 10.14806/ej.17.1.200.
- M. Bolger, M. Lohse, and B. Usadel, “Trimmomatic: a flexible trimmer for Illumina sequence data,” Bioinformatics, vol. 30, no. 15, pp. 2114–2120, Aug. 2014, doi: 10.1093/bioinformatics/btu170.
- Kim, G. Pertea, C. Trapnell, H. Pimentel, R. Kelley, and S. L. Salzberg, “TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions,” Genome Biol, vol. 14, no. 4, pp. R36–R36, Apr. 2013, doi: 10.1186/gb-2013-14-4-r36.
- Dobin et al., “STAR: ultrafast universal RNA-seq aligner,” Bioinformatics, vol. 29, no. 1, pp. 15–21, Jan. 2013, doi: 10.1093/bioinformatics/bts635.
- Dobin and T. R. Gingeras, “Mapping RNA-seq Reads with STAR,” Current Protocols in Bioinformatics, vol. 51, no. 1, pp. 11.14.1-11.14.19, Sep. 2015, doi: https://doi.org/10.1002/0471250953.bi1114s51.
- Wu and S. Nacu, “Wu TD, Nacu SFast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873-881,” Bioinformatics, vol. 26, pp. 873–881, Feb. 2010, doi: 10.1093/bioinformatics/btq057.
- Hu, H. Ge, M. Newman, and K. Liu, “OSA: a fast and accurate alignment tool for RNA-Seq,” Bioinformatics, vol. 28, no. 14, pp. 1933–1934, Jul. 2012, doi: 10.1093/bioinformatics/bts294.
- Wang et al., “MapSplice: accurate mapping of RNA-seq reads for splice junction discovery,” Nucleic Acids Res, vol. 38, no. 18, pp. e178–e178, Oct. 2010, doi: 10.1093/nar/gkq622.
- R. Rosenbloom et al., “The UCSC Genome Browser database: 2015 update,” Nucleic Acids Res, vol. 43, no. Database issue, pp. D670-81, 2015, doi: 10.1093/nar/gku1177.
- Dobin et al., “STAR: ultrafast universal RNA-seq aligner,” Bioinformatics, vol. 29, Oct. 2012, doi: 10.1093/bioinformatics/bts635.
- D. Jackman et al., “Tigmint: Correcting assembly errors using linked reads from large molecules,” BMC Bioinformatics, vol. 19, no. 1, Oct. 2018, doi: 10.1186/s12859-018-2425-6.
- Zhao, “Assessment of the impact of using a reference transcriptome in mapping short RNA-Seq reads,” PLoS One, vol. 9, no. 7, pp. e101374–e101374, Jul. 2014, doi: 10.1371/journal.pone.0101374.
- Chen et al., “Incorporating the human gene annotations in different databases significantly improved transcriptomic and genetic analyses,” RNA, vol. 19, Feb. 2013, doi: 10.1261/rna.037473.112.
- -Y. Wu, J. H. Phan, and M. D. Wang, “Assessing the impact of human genome annotation choice on RNA-seq expression estimates,” BMC Bioinformatics, vol. 14 Suppl 11, no. Suppl 11, pp. S8–S8, 2013, doi: 10.1186/1471-2105-14-S11-S8.
- D. Pruitt, T. Tatusova, and D. R. Maglott, “NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins,” Nucleic Acids Res, vol. 35, no. Database issue, pp. D61–D65, Jan. 2007, doi: 10.1093/nar/gkl842.
- Hsu, W. J. Kent, H. Clawson, R. M. Kuhn, M. Diekhans, and D. Haussler, “The UCSC Known Genes,” Bioinformatics, vol. 22, no. 9, pp. 1036–1046, May 2006, doi: 10.1093/bioinformatics/btl048.
- Flicek et al., “Ensembl 2014,” Nucleic Acids Res, vol. 42, no. Database issue, pp. D749–D755, Jan. 2014, doi: 10.1093/nar/gkt1196.
- Li and C. N. Dewey, “RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome,” BMC Bioinformatics, vol. 12, p. 323, Aug. 2011, doi: 10.1186/1471-2105-12-323.
- Trapnell et al., “Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation,” Nature Biotechnology, vol. 28, no. 5, pp. 511–515, 2010, doi: 10.1038/nbt.1621.
- Nicolae, S. Mangul, I. I. Măndoiu, and A. Zelikovsky, “Estimation of alternative splicing isoform frequencies from RNA-Seq data,” Algorithms Mol Biol, vol. 6, no. 1, p. 9, Apr. 2011, doi: 10.1186/1748-7188-6-9.
- Liao, G. K. Smyth, and W. Shi, “featureCounts: An efficient general-purpose program for assigning sequence reads to genomic features,” May 2013, doi: 10.1093/bioinformatics/btt656.
- Anders, P. T. Pyl, and W. Huber, “HTSeq–a Python framework to work with high-throughput sequencing data,” Bioinformatics, vol. 31, no. 2, pp. 166–169, Jan. 2015, doi: 10.1093/bioinformatics/btu638.
- Sims, I. Sudbery, N. E. Ilott, A. Heger, and C. P. Ponting, “Sequencing depth and coverage: key considerations in genomic analyses,” Nature Reviews Genetics, vol. 15, no. 2, pp. 121–132, 2014, doi: 10.1038/nrg3642.
- Anders and W. Huber, “Differential expression analysis for sequence count data,” Nature Precedings, 2010, doi: 10.1038/npre.2010.4282.1.
- Robinson and A. Oshlack, “Robinson MD, Oshlack A.. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25,” Genome Biol, vol. 11, p. R25, Mar. 2010, doi: 10.1186/gb-2010-11-3-r25.
- Robinson, D. Mccarthy, and G. Smyth, “Robinson MD, McCarthy DJ, Smyth GK.. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139-140,” Bioinformatics, vol. 26, pp. 139–140, Nov. 2009, doi: 10.1093/bioinformatics/btp616.
- s Cumbie et al., “GENE-Counter: A Computational Pipeline for the Analysis of RNA-Seq Data for Gene Expression Differences,” PLoS One, vol. 6, p. e25279, Oct. 2011, doi: 10.1371/journal.pone.0025279.
- Tarazona, F. García-Alcalde, J. Dopazo, A. Ferrer, and A. Conesa, “Differential expression in RNA-seq: a matter of depth,” Genome Res, vol. 21, no. 12, pp. 2213–2223, Dec. 2011, doi: 10.1101/gr.124321.111.
- Di, D. W. Schafer, J. S. Cumbie, and J. H. Chang, “The NBP Negative Binomial Model for Assessing Differential Gene Expression from RNA-Seq,” Statistical Applications in Genetics and Molecular Biology, vol. 10, no. 1, 2011, doi: doi:10.2202/1544-6115.1637.
- Trapnell et al., “Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks,” Nat Protoc, vol. 7, no. 3, pp. 562–578, Mar. 2012, doi: 10.1038/nprot.2012.016.
- O. Consortium, “Gene Ontology Consortium: going forward,” Nucleic Acids Res, vol. 43, no. Database issue, pp. D1049–D1056, Jan. 2015, doi: 10.1093/nar/gku1179.
- Kanehisa, S. Goto, S. Kawashima, Y. Okuno, and M. Hattori, “The KEGG resource for deciphering the genome,” Nucleic Acids Res, vol. 32, no. Database issue, pp. D277–D280, Jan. 2004, doi: 10.1093/nar/gkh063.
- -W. Yeh et al., “PSPC1 mediates TGF-β1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis,” Nature Cell Biology, vol. 20, no. 4, pp. 479–491, 2018, doi: 10.1038/s41556-018-0062-y.
- Hinz, “The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship,” Matrix Biology, vol. 47, pp. 54–65, 2015, doi: https://doi.org/10.1016/j.matbio.2015.05.006.
- Govinden and K. D. Bhoola, “Genealogy, expression, and cellular function of transforming growth factor-β,” Pharmacology & Therapeutics, vol. 98, no. 2, pp. 257–265, 2003, doi: https://doi.org/10.1016/S0163-7258(03)00035-4.
- LeClair and V. Lindner, “The Role of Collagen Triple Helix Repeat Containing 1 in Injured Arteries, Collagen Expression, and Transforming Growth Factor β Signaling,” Trends in Cardiovascular Medicine, vol. 17, no. 6, pp. 202–205, 2007, doi: https://doi.org/10.1016/j.tcm.2007.05.004.
- Hatzirodos et al., “Linkage of regulators of TGF- activity in the fetal ovary to polycystic ovary syndrome,” FASEB J, vol. 25, pp. 2256–2265, Mar. 2011, doi: 10.1096/fj.11-181099.
- Raja-Khan, M. Urbanek, R. J. Rodgers, and R. S. Legro, “The role of TGF-β in polycystic ovary syndrome,” Reproductive Sciences, vol. 21, no. 1. pp. 20–31, Jan. 2014. doi: 10.1177/1933719113485294.
- -Y. Liu et al., “Oncotarget 49897 www.impactjournals.com/oncotarget Ring finger protein 125, as a potential highly aggressive and unfavorable prognostic biomarker, promotes the invasion and metastasis of human gallbladder cancers via activating the TGF-β1-SMAD3-ID1 signaling pathway,” 2017. [Online]. Available: www.impactjournals.com/oncotarget/
- S. Saha, G. Caviness, G. Yi, E. L. Raymond, M. L. Lamine Mbow, and C. Cheng Kao, “E3 Ubiquitin Ligase RNF125 Activates Interleukin-36 Receptor Signaling and Contributes to Its Turnover,” Journal of Innate Immunity, vol. 10, no. 1, pp. 56–69, Feb. 2018, doi: 10.1159/000481210.
- Lin, W. Xing, Y. Li, Y. Xie, X. Tang, and Q. Zhang, “Downregulation of serum long noncoding RNA GAS5 may contribute to insulin resistance in PCOS patients,” Gynecological Endocrinology, vol. 34, no. 9, pp. 784–788, Sep. 2018, doi: 10.1080/09513590.2018.1459548.
- yuan Zhang, F. fan Zhu, Y. jun Zhu, Y. jing Hu, and X. Chen, “Effects of IL-18 on the proliferation and steroidogenesis of bovine theca cells: Possible roles in the pathogenesis of polycystic ovary syndrome,” Journal of Cellular and Molecular Medicine, vol. 25, no. 2, pp. 1128–1139, Jan. 2021, doi: https://doi.org/10.1111/jcmm.16179.
- Kordowitzki, G. Sokołowska, M. Wasielak-Politowska, A. Skowronska, and M. T. Skowronski, “Pannexins and connexins: Their relevance for oocyte developmental competence,” International Journal of Molecular Sciences, vol. 22, no. 11. MDPI, Jun. 01, 2021. doi: 10.3390/ijms22115918.
- Iwamoto et al., “Pannexin 3 regulates intracellular ATP/cAMP levels and promotes chondrocyte differentiation,” Journal of Biological Chemistry, vol. 285, no. 24, pp. 18948–18958, Jun. 2010, doi: 10.1074/jbc.M110.127027.
- Singla, Y. Gupta, M. Khemani, and S. Aggarwal, “Thyroid disorders and polycystic ovary syndrome: An emerging relationship,” Indian Journal of Endocrinology and Metabolism, vol. 19, no. 1. Medknow Publications, pp. 25–29, Jan. 01, 2015. doi: 10.4103/2230-8210.146860.
- I. Muderris, A. Boztosun, G. Oner, and F. Bayram, “Effect of thyroid hormone replacement therapy on ovarian volume and androgen hormones in patients with untreated primary hypothyroidism,” Ann Saudi Med, vol. 31, no. 2, pp. 145–151, 2011.
- E. van den Boogaard et al., “Significance of (sub) clinical thyroid dysfunction and thyroid autoimmunity before conception and in early pregnancy: a systematic review,” Hum Reprod Update, vol. 17, no. 5, pp. 605–619, 2011.
Publication History
Submitted: April 04, 2023
Accepted: April 20, 2023
Published: May 01, 2023
Identification
D-0114
Citation
Pradip Rijal, Aatiqa Tariq & Syeda Hajra Batool (2023). The Study of Differential Expression of Genes Controlling Reproductive Function in Immune Cells of PCOS Women. Dinkum Journal of Medical Innovations, 2(05):157-169.
Copyright
© 2023 DJMI. All rights reserved