

")
Publication History
Submitted: April 02, 2023
Accepted: April 20, 2023
Published: May 01, 2023
Identification
D-0115
Citation
Pradip Rijal (2023). Advances of NGS in Understanding of Epilepsy Genetics and Recent Discoveries of Gene in Monogenic Epilepsies. Dinkum Journal of Medical Innovations, 2(05):170-181.
Copyright
© 2023 DJMI. All rights reserved
170-181
Advances of NGS in Understanding of Epilepsy Genetics and Recent Discoveries of Gene in Monogenic EpilepsiesReview Article
Pradip Rijal 1*
- Sunsari Technical College, Nepal; rijal.pradipn@gmail.com
* Correspondence: rijal.pradipn@gmail.com
Abstract: A group of neurological conditions with varying disease patterns that affect individuals of all ages and genders is known as epilepsy. It might show up as sudden sensory problems, loss of consciousness, aberrant autonomic nerve function, paroxysmal movement, and mental issues. It is distinguished by the vulnerability to both the absence of seizures and the development of long-term seizures. Next-generation sequencing technology has completely changed the way genes in monogenic epilepsies are identified over the past ten years. Improvements in our understanding of the underlying genetic variations that cause genetic epilepsies have had a significant impact on both the downstream personalized treatment strategy and the diagnosis of genetic epilepsies. This review article aims to give a general overview of monogenic epilepsy, highlight the ways that Next Generation Sequencing technology has improved our understanding of the genetics of the condition, and go through some of the precision medicine strategies for treating monogenic epilepsy.
Keywords: advances, epilepsy, genetics, gene, monogenic epilepsies
- INTRODUCTION
A group of neurological conditions with varying disease patterns that affect individuals of all ages and genders is known as epilepsy [1]. It might show up as sudden sensory problems, loss of consciousness, aberrant autonomic nerve function, paroxysmal movement, and mental issues. It is distinguished by the vulnerability to both the absence of seizures and the development of long-term seizures [2]. Although epilepsy has a significant lack of heredity [3], it is frequently seen as a heritable disorder [4, 5]. Additionally, research on the epidemiology of twins and families provide encouraging proof that epilepsy is inherited [6, 7]. The causes of epilepsy are diverse, much like those of many other neurological conditions. Most occurrences of epilepsy have been related to genetic origins [8], although certain cases have been connected to underlying conditions such brain trauma, tumours, and strokes, as well as chronic infectious aetiologies like HIV, syphilis, and neurocysticercosis [9]. In terms of genetics, epilepsy falls into two basic categories: i) Primary Epilepsy: genes and loci related ii) Secondary Epilepsy:Genes linked to neurological conditions in which epilepsy may be a symptom8. There are two primary genetic causes of epilepsy: one is monogenic, and the other is polygenic. While polygenic types of epilepsy involve variations in numerous genes, monogenic variants arise from mutations in a single gene. Around 40–50 million people worldwide suffer from active epilepsy (primary and secondary) [10], with nearly 80 percent of these persons living in developing nations [11]. When comparing the annual incidence of epilepsy in developing and western countries, there is a notable difference. In contrast to underdeveloped countries, where the maximum incidence is 187/100,000, western countries have comparatively low incidences, ranging from 33.3 to 82/100,000 [12, 13]. One hundred fifty thousand new instances of epilepsy are identified in the United States alone, affecting 2.2 million people [1]. Figure 1 below illustrates the prevalence of epilepsy throughout several global regions, highlighting the condition’s burden on society.
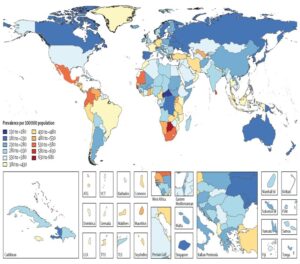
Figure 1: Prevalence of epilepsy in different parts of the world
Epileptic encephalopathy typically manifests as developmental delays encompassing a range of neurological and non-neurological diseases, and it usually begins in childhood. West syndrome, Lennox-Gastaut syndrome, Dravet syndrome, atypical benign partial epilepsy, electrical status epilepticus during sleep, syndrome linked to febrile infection-related epilepsy, Landau-Kleffner syndrome, and migrating partial seizures of infancy are among the severe forms of childhood epilepsies. It has both the incidence and prevalence into adulthood, despite the fact that it has long been thought of as a disease affecting only children. According to estimates, 10.8 out of 1000 older people have epilepsy, with a 2.4 annual incidence [14]. The worldwide burden of disease is largely attributed to the prevalence of epilepsy and seizures in the senior population. It’s interesting to note that after the age of 60, around 25% of cases of newly onset epilepsy occur [15]. Epilepsy is one of the most prevalent nervous system disorders in the aged population, coming in second only to dementia and stroke [16]. Indeed, the onset of epilepsy and seizures is most likely to occur in old age [17]. Genetic abnormalities of many kinds are linked to epilepsy and seizure proneness. For example, epilepsy and chromosomal anomalies have been associated. Wolf-Hirschhorn syndrome [18], ring 20 chromosome syndrome [19], Miller-Dieker syndrome [20], 1p36 monosomy [21], and 18q-syndrome [21] are a few examples of chromosomal abnormality linked to epilepsy. In a similar vein, copy number variation, or CNV, is important in the development of clinical epilepsy [22]. It has also been determined that epilepsy is caused by a mutation in the mitochondrial DNA [23]. Furthermore, a number of epigenetic variables, such as transcriptional regulation [25, 26], histone modification [24], and DNA methylation [25], are linked to epilepsy.
- LITERATURE REVIEW
As the name suggests, a single faulty gene is the predictor of monogenic epilepsy. Only 1% to 2% of people have monogenic epilepsies [27, 28]. Monogenic epilepsies include both monogenic severe epilepsies and monogenic family epilepsies, which are collectively referred to as epileptic encephalopathies (EE) [29]. Monogenic origins of hereditary epilepsies account for 40% of cases of severe epilepsies in humans [30]. Since the clinical significance of many common and rare epilepsy is still unknown due to their highly heterogeneous genetic architecture, these causes have received a great deal of attention for clinical diagnostic purposes as well as gene discovery strategies, novel therapeutic modalities, and possibly even epilepsy prevention [31]. Idiopathic or genetic generalised epilepsies are a subgroup of monogenic epilepsy characterised by a form of seizure that depresses consciousness and causes disruptions to both sides of the brain’s activity. These epilepsies are the most common and can make up up to one-third of all epilepsies [32]. Focal epilepsies, another kind of monogenic epilepsies also known as partial seizure, account for 60% of all epilepsies [33]. Focal epilepsies were once believed to be acquired diseases. However, an increasing body of evidence indicates that focused epilepsies are inherited rather than acquired [34]. Even though focal seizures typically only impact one area of the brain, they can sometimes begin in one region and spread to the other. A group of severe forms of monogenic epilepsies known as epileptic encephalopathy (EE) and developmental and epileptic encephalopathy (DEE) are characterised by early onset of frequent seizures, resistance to medication, and extremely abnormal electroencephalograms (EEGs), which ultimately cause intellectual disability and developmental delay. The terms “EE” and “DEE” have different conceptual meanings. For example, the term “emergence” (EE) refers to a hypothesised reciprocal interaction in which developmental delay and epilepsy are thought to be caused by one another [35]. EEs and DEEs are linked to a number of age-related epileptic encephalopathy disorders. Early myoclonic encephalopathy, Ohtahara syndrome, or migrating partial seizures in the neonatal period are among the syndromes classified as malignant epilepsies [36]. Two more syndromes linked to newborns are West and Dravet [37, 38]. Similarly, Landau-Kleffner and juvenile myoclonic adolescence syndrome begin in individuals between the ages of 18 months and 13 years and adolescence, respectively, [40]. Lennox-Gastaut syndrome is a severe epilepsy that begins in childhood [39]. For example, familial generalised epilepsies are caused by mutations in the genes GABRA1, GABRA2, SCN1A, and SCN1B [41–44]. For nonfamilial genetic generalised epilepsies, on the other hand, recurrent micro deletions at 15q13.3, 16p13.11, and 15q11.2 were discovered to be the causal culprits [45–47]. It is known that many focal epilepsy syndromes have a hereditary basis. For instance, mutations in (CHRNA4, CHRNA2, CHRNB2) produce autosomal dominant sleep-related hypermotor epilepsy (ADSHE), a monogenic non-acquired focal epilepsy [48-51]. Similarly, a mutation in the genes LGI [52] and RELN [53] results in autosomal dominant epilepsy with auditory characteristics (ADEAF), another familial focal epilepsy condition. Focal epilepsy, often referred to as autosomal dominant Rolandic epilepsy with speech dyspraxia, is brought on by a mutation in the GRIN2A gene [54]. It has been observed that benign family neonatal epilepsy is caused by mutations in the KCNQ2, KCNQ3, and KCNQ3 genes [55, 56]. Benign familial neonatal infantile epilepsy is caused by mutations in the genes. The genes (DEPDC5, NPRL2, NPRL3) that have been linked to the pathophysiology of familial focal epilepsy with variable foci 8FFEVF are [57]. Furthermore, familial mesial temporal lobe epilepsy (FMTLE) has also been linked to mutations in DEPDC5, however these are not common causes [58]. In other words, the genotype-phenotype association in monogenic epilepsies is not as direct and obvious as it is in many other genetic conditions. Familial epilepsies and epileptic encephalopathies rank at opposite extremes of the severity spectrum; yet, they can both be generated by the same gene, resulting in benign and sever epilepsy phenotypes. We give a few instances of this occurrence below. The potassium ion channel gene, or KCNQ2 gene, functions as a tiny gate on the surface of brain cells, allowing potassium ions to leave when the cell becomes overly active. Monogenic epilepsy is characterised by a mutation in KCNQ2 on chromosome 20q, which is like having a harder brake [59]. It has been discovered that KCNQ2 mutations not only result in severe neonatal epileptic encephalopathies [61], but also benign familial neonatal seizures [60]. One of the genes linked to voltage-gated sodium channels, SCN1A, encodes the Nav1.1 alpha subunits of sodium channels. These channels are mostly found in the brain, where they regulate the flow of sodium ions to the cells [62]. The resting potential as well as the production and transmission of action potentials in neurons—which aid in deciding when to release neurotransmitters—are dependent on these channels [63]. A variety of diseases, including febrile seizures, generalised epilepsy with febrile seizures [64], and the severe kind of epilepsy known as Dravet syndrome [65], are brought on by mutations in this gene. However, genetic backgrounds have a significant influence on the severity of Dra-vets syndrome [66, 67]. PCDH19, a gene identified in Xp22.1, encodes protocadherin-19, a calcium-dependent cell adhesion molecule that is known to have a variety of roles in migration, the creation of neural circuits, and the regulation of neuronal progenitor proliferation [70–72]. Female epilepsy caused by mutations in the PCDH19 gene is strikingly associated with cognitive impairment [73, 74]. Nonetheless, patients with a mutation in the PCDH19 gene, which primarily affects females, exhibit a phenotype similar to that of a Dravet [75]. Moreover, mutations in the GABRA1 and STXBP1 genes have been linked to the Dravet-like phenotype [76]. The calcium-activated potassium channels (KCNT1 gene, chromosome 9) are widely expressed in the nervous system and are essential for the transport of potassium ions out of cells, which produces and transmits electrical current that excites neurons and sends signals to the brain [78, 79]. Two different forms of epilepsy have been linked to mutations in KCNT1: epileptic encephalopathy malignant migrating focal seizures of infancy (MMFSI) [81] and familial focal epilepsy syndrome, also known as autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) [80]. Furthermore, autosomal dominant pathogenic mutations in KCNT1 have also been linked to the illnesses known as Ohtahara and West syndromes [82, 83]. The over three billion base pairs that make up the human genome are home to all the secrets of human existence. Through the sequencing of these billions of base pairs, the Hu-man Genome Project is the first scientific method that has contributed to the unravelling of the enigma of life [84]. Numerous conventional methods, such as genome-wide association studies and linkage analysis, are primarily concerned with inherited genetic variation. Using genomics and related technologies has improved our current understanding of biology. Targeted gene panels, whole exome sequencing, and whole genome sequencing are examples of next-generation sequencing technologies that have demonstrated a tremendous advancement in our understanding of biological phenomena that are intricately linked. Numerous novel epilepsy genes, including those causing de novo mutations, generalised and localised epilepsy syndromes, and other familial epilepsies, have been found thanks to NGS-based genomic research. We are able to effectively investigate the function of de novo mutations and their impact to health and disease through whole-genome and whole-exome sequencing techniques. Even though they are uncommon on their own, these mutations offer a way to describe the heritability of complicated genetic illnesses that may not be achievable with conventional methods [85]. Due to a mutation in the SCNA1 gene, Dravet Syndrome was not detected by traditional Sanger sequencing. But focused gene panel testing has shown to be helpful in determining SCN1A’s genetic contribution to Dravet syndrome [86], supporting the value of gene panel testing in treatment strategies and predicting the course and prognosis of disease. Through whole-exome sequencing and targeted resequencing, it was discovered that not only SCNA1, but also GABRA1 and STXBP1 were significant contributors as novel genes for Dravet Syndrome. This suggests that successful gene identification with Dravets syndrome is possible without a molecular diagnosis, opening a wide door of therapeutic approach through NGS [87]. Similar to this, NGS discovered two de novo and one maternal pathogenic variant of CHD2, which was reported as a novel loss-of-function mutation and causes epileptic encephalopathy that manifests in children [88]. Targeted next-generation sequencing revealed many mutations in the DEPDC5 gene, including those associated with focal epilepsy with febrile seizures and cortical development malformation [89]. A case of homozygous DEPDC5 mutation-related localised cortical dysplasia was found in the same investigation. Furthermore, in 11 families with mild focal epilepsies, eight heterozygous mutations were found; among these, 13 patients in eight families with focal epilepsy and febrile episodes had the mutation. The most popular use of next-generation sequencing is exome sequencing. Trio sequencing, which involves sequencing the genomes of the epileptic person and both parents, has proven to be an effective method of discovering novel risk factors for epileptic encephalopathies [90, 91]. Trio-based analysis was used, for example, to identify the genes ALG1392, GABRB392, HCN193, GRIN2A94, GABRA195, GNAO196, SCN2A97, SCN8A98, and SLC35A2 [99]. Furthermore, three de novo heterozygous missense mutations in the DNM1 gene were found by exome sequencing in a 15-year-old girl with developmental and epileptic encephalopathy whose parents were unaffected [100]. The findings that mutations in DNM1 impair synaptic vesicle endocytosis [101] and cause severe epileptic encephalopathy were consistent with all of the mutations. This early infantile epileptic encephalopathy cohort was treated with whole genome sequencing and thorough variant finding methods without any prior diagnosis. The identification of a likely pathogenic mutation in every subject is a notable illustration of how whole genome sequencing can be applied to speed up and lower the cost of clinical diagnosis of EIEE [102]. It also emphasises the superiority of WES as a diagnostic technique for EIEE and related genetic traits. The discovery of de novo mutations in the genes of probable clinical significance to epilepsy (KCNH5, CLCN4, and AGHGEF15) carried by children with early infantile epileptic encephalopathy [103] indicates that WES is crucial for the molecular genetic diagnosis of sporadic epilepsies in children, especially for difficult-to-control early-onset seizures. The most promising targets for precision therapy in monogenic epilepsy have been identified thanks to genetic discoveries in epilepsy, which have been fueled by advances in sequencing technologies [104]. It may only address a portion of the true functional abnormalities and is still only appropriate to a small percentage of patients with monogenic epilepsies [105]. A few precision medicine strategies for treating monogenic epilepsies are covered here. As was previously mentioned, Dravet syndrome is a severe form of epilepsy that develops in childhood due to a loss-of-function mutation of the gene SCNA1, which encodes the neuronal voltage-gated sodium channel alpha subunit Nav1.1. This mutation reduces inhibitory neurotransmission and causes brain hyperexcitability [106] because it prevents GABAergic inhibitory neurons from firing action potentials. It was discovered that giving Hm1a intracerebroventricular infusion prevents seizures and early death in rats with Dravet syndrome [107]. The use of precision medicine offers hope for the treatment of such uncontrollably severe epilepsy. For the first time, a clinical trial demonstrates the effectiveness of cannabidiol in treating drug-resistant seizures in patients with Dravet syndrome [108]. Similarly, sodium channel blockers are not advised for SCN1A mutations because these mutations are loss of function mutations and further blocking of sodium channels increases seizures in Dravet patients [110, 111]. However, fenfluramine, a serotonin releasing drug, has recently been licenced as therapy for seizures associated with Dravet syndrome [109]. One of the most prevalent monogenic epilepsy syndromes is PRRT2. A mutation in PRRT2 results in hemiplegic migraine, paroxysmal dyskinesia, and familial infantile seizures, among other disorders112. Furthermore, mutations causing loss of function in PPRT2113 result in paroxysmal neurological diseases. Studies have demonstrated that PRRT2 deficiency causes sodium channel hyperactivity in mouse and human neurons [114], highlighting the potential use of sodium channel blockers as a targeted treatment. The efficiency of sodium blockers such as levetiracetam and lacosamide in treating children with benign infantile epilepsy further supports this [115]. Focused epilepsy with varying foci is caused by a frameshift mutation in the DEPDC5 gene, according to linkage analysis and exome sequencing [116]. Genetic focal epilepsy is frequently linked to the mechanistic target of rapamycin complex 1 (mTORC1) hyperactivation, which is caused by a loss of function mutation in DEPDC5 [117]. For example, it has been demonstrated that rapamycin inhibits mTORC1 to prevent seizures and early death in DEPD5 knockout mice [118]. Similarly, it was discovered that long-term mTORC1 inhibition might restore the biochemical and behavioural abnormalities brought on by the loss of neuronal Depdc5 in mice [119]. Furthermore, research has demonstrated that the ketogenic diet inhibits the mTOR (mammalian target of rapamycin). The paradigm shift in genomic science from data generation to data interpretation has been brought about by next-generation sequencing technology. Prior to the development of NGS, the area of genomics lacked resources, yet a vast amount of data has been produced. The interpretation of data is currently the main difficulty in the field of genomics. We must carefully analyse the data using a sophisticated statistical method in order to prevent making incorrect claims about the genes that cause certain phenotypes and their causal relationships. Research on genetics in epilepsies was inconsistent with other areas of neurobiology, such as studies on autism and schizophrenia. The intricacy of the genotype and phenotype was the main cause of this. Up till now, a wide range of genetic abnormalities that contribute to the epilepsy condition have been found through the extensive use of NGS techniques. This demonstrates unequivocally that NGS has emerged as the best practical choice for the standard diagnosis of epilepsy. The epilepsies are genuinely at the forefront of their genetic revolution thanks to the large-scale genomic analyses that NGS has made possible. Still, clinical and research labs need to think about clinical utility, diagnostic yield, and cost effectiveness. A de novo mutation does not always imply that the disease manifestation is caused by the recently discovered mutation. More functional research, the discovery of individuals who share the same gene mutations and are similarly affected, or the use of a sizable sample size may be required in order to confirm the genetic origin of a novel variant. It will become more difficult to distinguish between causative and non-causative variation as the volume of data produced by massively parallel sequencing increases. Thus, in order to maximise the likelihood of discovering accidental variations, we must carefully choose the epilepsy genes. Certain genes can express themselves at any time during life, from early childhood to adulthood, depending on a variety of cellular and environmental cues. As a result, there are many fascinating questions in this area. Whether current genomic technologies are able to accurately measure every qualitative aspect and pinpoint the causative variations linked to the illness phenotypes is still an open question.
- CONCLUSION
The identification of genes in monogenic epilepsies has been transformed in the past ten years by next-generation sequencing methods. The diagnosis of genetic epilepsies and the downstream personalized medicine strategy have both benefited greatly from our growing understanding of the causative genetic variations. This review paper aims to give a general overview of monogenic epilepsy, highlight some of the precision medicine approaches for treating monogenic epilepsy, and outline how Next Generation Sequencing technology has improved our understanding of the genetics of monogenic epilepsy.
REFERENCES
- England, M. J., Liverman, C. T., Schultz, A. M. & Strawbridge, L. M. Epilepsy across the spectrum: Promoting health and understanding.: A summary of the Institute of Medicine report. Epilepsy & Behavior 25, 266–276 (2012).
- Panayiotopoulos, C. P. Treatment of Typical Absence Seizures and Related Epileptic Syndromes. Paediatric Drugs 3, 379–403 (2001).
- Speed, D. et al. Describing the genetic architecture of epilepsy through heritability analysis. Brain 137, 2680–2689 (2014).
- Bourgeois, B. F., Dodson, E., Nordli Jr, D. R., Pellock, J. M. & Sankar, R. Pediatric epilepsy: diagnosis and therapy. (Demos Medical Publishing, 2007).
- Lynch, D. R. Neurogenetics: scientific and clinical advances. (CRC Press, 2005).
- Berkovic, S. F., Howell, R. A., Hay, D. A. & Hopper, J. L. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol 43, 435–445 (1998).
- Steinlein, O. K. Genes and mutations in human idiopathic epilepsy. Brain and Development 26, 213–218 (2004).
- Poduri, A. & Lowenstein, D. Epilepsy genetics–past, present, and future. Curr Opin Genet Dev 21, 325–332 (2011).
- Ngarka, L., Siewe Fodjo, J. N., Aly, E., Masocha, W. & Njamnshi, A. K. The Interplay Between Neuroinfections, the Immune System and Neurological Disorders: A Focus on Africa. Front Immunol 12, 803475 (2022).
- Maryam Majeed, Rubab Ali, Umm-e-Aimen & Aqsa Jabeen. Medical Staff, Workplace Bullying and Its Effects on the Performance. Dinkum Journal of Medical Innovations, 2(04):120-125.
- Marie Diack. Factors Influencing Relational Violence in Nepali Married Relationships. Dinkum Journal of Medical Innovations, 2(04):126-133.
- Gul Rukh Malik, Rubab Zahra & Tahir Rana. Autism Spectrum Disorders Frequency in Asian Countries. Dinkum Journal of Medical Innovations, 2(04):134-139.
- Parshu Ram Chaudhary, Asbin Bandhari & Parshu Kirby. Aeroallergens and Significant Environmental Pollutants: Aeroallergen Sensitivity Symptoms. Dinkum Journal of Medical Innovations, 2(04):140-149.
- Imran Mehfooz Khan, Aron Kumar & Pradeep Sahejpal. Extensive Management Medical Incidents Provided to Gallbladder Cancer Using Cholecystectomy Specimen. Dinkum Journal of Medical Innovations, 2(04):150-156.
- Brodie, M. J., Elder, A. T. & Kwan, P. Epilepsy in later life. The Lancet Neurology 8, 1019–1030 (2009).
- Hesdorffer, D. C. et al. Estimating risk for developing epilepsy: a population-based study in Rochester, Minnesota. Neurology 76, 23–27 (2011).
- Kagitani-Shimono, K. et al. Epilepsy in Wolf-Hirschhorn Syndrome (4p-). Epilepsia 46, 150–155 (2005).
- Atkins, L., Miller, W. L. & Salam, M. A ring-20 chromosome. J Med Genet 9, 377–380 (1972).
- Singh, R., McKinlay Gardner, R. J., Crossland, K. M., Scheffer, I. E. & Berkovic, S. F. Chromosomal abnormalities and epilepsy: a review for clinicians and gene hunters. Epilepsia 43, 127–140 (2002).
- Shaffer, L. G. & Heilstedt, H. A. Terminal deletion of 1p36. The Lancet 358, S9 (2001).
- Olson, H. et al. Copy number variation plays an important role in clinical epilepsy. Ann Neurol 75, 943–958 (2014).
- RAHMAN, S. Mitochondrial disease and epilepsy. Developmental Medicine & Child Neurology 54, 397–406 (2012).
- Liu, X. et al. New differentially expressed genes and differential DNA methylation underlying refractory epilepsy. Oncotarget 7, 87402–87416 (2016).
- Hwang, J.-Y., Aromolaran, K. A. & Zukin, R. S. Epigenetic Mechanisms in Stroke and Epilepsy. Neuropsychopharmacology 38, 167–182 (2013).
- Jagirdar, R., Drexel, M., Kirchmair, E., Tasan, R. & Sperk, G. Rapid changes in expression of class I and IV histone deacetylases during epileptogenesis in mouse models of temporal lobe epilepsy. Exp Neurol (2015) doi:10.1016/j.expneurol.2015.07.026.
- Avanzini, G. Of Cabbages and Kings: Do We Really Need a Systematic Classification of Epilepsies? Epilepsia 44, 12–13 (2003).
- Stafstrom, C. E. & Carmant, L. Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb Perspect Med 5, a022426 (2015).
- Helbig, I. & Lowenstein, D. H. Genetics of the epilepsies: where are we and where are we going? Curr Opin Neurol 26, 179–185 (2013).
- Guerrini, R., Balestrini, S., Wirrell, E. C. & Walker, M. C. Monogenic Epilepsies. Neurology 97, 817 (2021).
- Tlusta, E. et al. Clinical relevance of patients with epilepsy included in clinical trials. Epilepsia 49, 1479–1480 (2008).
- Jallon, P., Loiseau, P. & Loiseau, J. Newly diagnosed unprovoked epileptic seizures: presentation at diagnosis in CAROLE study. Coordination Active du Réseau Observatoire Longitudinal de l’ Epilepsie. Epilepsia 42, 464–475 (2001).
- Devinsky, O. et al. Nature Reviews Disease Primers 4, 18024 (2018).
- Thomas, R. H. & Berkovic, S. F. The hidden genetics of epilepsy—a clinically important new paradigm. Nature Reviews Neurology 10, 283–292 (2014).
- Specchio, N. & Curatolo, P. Developmental and epileptic encephalopathies: what we do and do not know. Brain 144, 32–43 (2021).
- Yamamoto, H., Okumura, A. & Fukuda, M. Epilepsies and epileptic syndromes starting in the neonatal period. Brain and Development 33, 213–220 (2011).
- D’Alonzo, R., Rigante, D., Mencaroni, E. & Esposito, S. West Syndrome: A Review and Guide for Paediatricians. Clinical Drug Investigation 38, 113–124 (2018).
- Anwar, A., Saleem, S., Patel, U. K., Arumaithurai, K. & Malik, P. Dravet Syndrome: An Overview. Cureus 11, e5006–e5006 (2019).
- Jahngir, M. U., Ahmad, M. Q. & Jahangir, M. Lennox-Gastaut Syndrome: In a Nutshell. Cureus (2018) doi:10.7759/cureus.3134.
- Jahngir, M. U., Ahmad, M. Q. & Jahangir, M. Lennox-Gastaut Syndrome: In a Nutshell. Cureus (2018) doi:10.7759/cureus.3134.
- Escayg, A., MacDonald T, B. & Meisler H, M. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 24, 343–345 (2000).
- Harkin A, L., Bowser N, D. & Dibbens M, L. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet 70, 530–536 (2002).
- Wallace H, R., Wang W, D. & Singh, R. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet 19, 366–370 (1998).
- Baulac, S., Huberfeld, G. & Gourfinkel-An, I. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet 28, 46–48 (2001).
- Helbig, I., Mefford C, H. & Sharp J, A. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet 41, 160–162 (2009).
- Dibbens M, L., Mullen, S. & Helbig, I. Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Hum Mol Genet 18, 3626–3631 (2009).
- de Kovel G, C., Trucks, H. & Helbig, I. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain 133, 23–32 (2010).
- Steinlein, O. K. et al. A missense mutation in the neuronal nicotinic acetylcholine receptor α4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 11, 201–203 (1995).
- Steinlein, O. K., Hoda, J.-C., Bertrand, S. & Bertrand, D. Mutations in familial nocturnal frontal lobe epilepsy might be associated with distinct neurological phenotypes. Seizure 21, 118–123 (2012).
- Steinlein, O. K., Hoda, J.-C., Bertrand, S. & Bertrand, D. Mutations in familial nocturnal frontal lobe epilepsy might be associated with distinct neurological phenotypes. Seizure 21, 118–123 (2012).
- Bisulli, F., Licchetta, L. & Tinuper, P. Sleep related hyper motor epilepsy (SHE): a unique syndrome with heterogeneous genetic etiologies. Sleep Science and Practice 3, (2019).
- Senechal, K., Thaller, C. & Noebels, J. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet 14, 1613–1620 (2005).
- Michelucci, R. & Nobile, C. Autosomal Dominant Epilepsy with Auditory Features Synonyms: ADEAF, Autosomal Dominant Lateral Temporal Epilepsy (ADLTE). (1993).
- Carvill, G. L. et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet 45, 1073–1076 (2013).
- Goldberg-Stern, H., Kaufmann, R., Kivity, S., Afawi, Z. & Heron, S. E. Novel Mutation in <em>KCNQ2</em> Causing Benign Familial Neonatal Seizures. Pediatric Neurology 41, 367–370 (2009).
- Heron, S. E. et al. Deletions or duplications in KCNQ2 can cause benign familial neonatal seizures. J Med Genet 44, 791–796 (2007).
- Weckhuysen, S. et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia 57, 994–1003 (2016).
- Striano, P. et al. DEPDC5 mutations are not a frequent cause of familial temporal lobe epilepsy. Epilepsia 56, e168–e171 (2015).
- Hani, A. J. & Mikati, M. A. 70 – Electroclinical Syndromes: Neonatal Onset. in Swaiman’s Pediatric Neurology (Sixth Edition) (eds. Swaiman, K. F. et al.) 552–556 (Elsevier, 2017). doi:https://doi.org/10.1016/B978-0-323-37101-8.00070-9.
- Christian, B. et al. A Potassium Channel Mutation in Neonatal Human Epilepsy. Science (1979) 279, 403–406 (1998).
- Weckhuysen, S. et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Annals of Neurology 71, 15–25 (2012).
- Noda, M. & Hiyama, T. Y. Sodium sensing in the brain. Pflugers Archiv : European journal of physiology 467, 465–474 (2015).
- Wang, J., Ou, S.-W. & Wang, Y.-J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels (Austin) 11, 534–554 (2017).
- Goldberg-Stern, H. et al. Broad Phenotypic Heterogeneity due to a Novel SCN1A Mutation in a Family With Genetic Epilepsy With Febrile Seizures Plus. Journal of Child Neurology 29, 221–226 (2013).
- Damiano, J. A. et al. SCN1A Variants in vaccine-related febrile seizures: A prospective study. Annals of Neurology 87, 281–288 (2020).
- Yu, F. H. et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 9, 1142–1149 (2006).
- Genton, P., Velizarova, R. & Dravet, C. Dravet syndrome: The long-term outcome. Epilepsia 52, 44–49 (2011).
- Wolverton, T. & Lalande, M. Identification and characterization of three members of a novel subclass of protocadherins. Genomics 76, 66–72 (2001).
- Cooper, S. R., Jontes, J. D. & Sotomayor, M. Structural determinants of adhesion by Protocadherin-19 and implications for its role in epilepsy. Elife 5, e18529 (2016).
- Cooper, S. R. et al. Protocadherins control the modular assembly of neuronal columns in the zebrafish optic tectum. Journal of Cell Biology 211, 807–814 (2015).
- Biswas, S., Emond, M. R. & Jontes, J. D. Protocadherin-19 and N-cadherin interact to control cell movements during anterior neurulation. Journal of Cell Biology 191, 1029–1041 (2010).
- Fujitani, M., Zhang, S., Fujiki, R., Fujihara, Y. & Yamashita, T. A chromosome 16p13. 11 microduplication causes hyperactivity through dysregulation of miR-484/protocadherin-19 signaling. Mol Psychiatry 22, 364–374 (2017).
- Dibbens, L. M. et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 40, 776–781 (2008).
- Camacho, A. et al. Cognitive and behavioral profile in females with epilepsy with PDCH19 mutation: two novel mutations and review of the literature. Epilepsy & Behavior 24, 134–137 (2012).
- Depienne, C. et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet 5, e1000381–e1000381 (2009).
- Carvill, G. L. et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology 82, 1245–1253 (2014).
- Wei, A. D. et al. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev 57, 463–472 (2005).
- Bhattacharjee, A. & Kaczmarek, L. K. For K+ channels, Na+ is the new Ca2+. Trends Neurosci 28, 422–428 (2005).
- Brown, M. R. et al. Amino‐termini isoforms of the Slack K+ channel, regulated by alternative promoters, differentially modulate rhythmic firing and adaptation. J Physiol 586, 5161–5179 (2008).
- Heron, S. E. et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 44, 1188–1190 (2012).
- Barcia, G. et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 44, 1255–1259 (2012).
- Martin, H. C. et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet 23, 3200–3211 (2014).
- Fukuoka, M. et al. Quinidine therapy for West syndrome with KCNTI mutation: a case report. Brain and Development 39, 80–83 (2017).
- Green, E. D., Watson, J. D. & Collins, F. S. Human Genome Project: Twenty-five years of big biology. Nature 526, 29–31 (2015).
- Veltman, J. A. & Brunner, H. G. De novo mutations in human genetic disease. Nature Reviews Genetics 13, 565–575 (2012).
- Jiwon Lee, Chung Lee, Woong-Yang Park & Jeehun Lee. Genetic Diagnosis of Dravet Syndrome Using Next Generation Sequencing-Based Epilepsy Gene Panel Testing. Ann Clin Lab Sci 50, 625–637 (2020).
- Carvill, G. L. et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology 82, 1245–1253 (2014).
- Wang, X. et al. Novel Loss-of-Function Variants in CHD2 Cause Childhood-Onset Epileptic Encephalopathy in Chinese Patients. Genes (Basel) 13, 908 (2022).
- Liu, L. et al. DEPDC5 Variants Associated Malformations of Cortical Development and Focal Epilepsy With Febrile Seizure Plus/Febrile Seizures: The Role of Molecular Sub-Regional Effect. Front Neurosci 14, 821 (2020).
- Investigators, E. and E. De novo mutations in the classic epileptic encephalopathies. Nature 501, 217 (2013).
- Appenzeller, S. et al. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. The American Journal of Human Genetics 95, 360–370 (2014).
- Investigators, E. and E. De novo mutations in the classic epileptic encephalopathies. Nature 501, 217 (2013).
- Nava, C. et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat Genet 46, 640–645 (2014).
- Carvill, G. L. et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet 45, 1073–1076 (2013).
- Carvill, G. L. et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology 82, 1245–1253 (2014).
- Nakamura, K. et al. De Novo mutations in GNAO1, encoding a Gαo subunit of heterotrimeric G proteins, cause epileptic encephalopathy. The American Journal of Human Genetics 93, 496–505 (2013).
- Ogiwara, I. et al. De novo mutations of voltage-gated sodium channel αII gene SCN2A in intractable epilepsies. Neurology 73, 1046–1053 (2009).
- Veeramah, K. R. et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. The American Journal of Human Genetics 90, 502–510 (2012).
- Kodera, H. et al. De Novo Mutations in SLC 35 A 2 Encoding a UDP‐Galactose Transporter Cause Early‐Onset Epileptic Encephalopathy. Hum Mutat 34, 1708–1714 (2013).
- Consortium, E.-R., Project, E. P. & Consortium, E. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 95, 360–370 (2014).
- Dhindsa, R. S. et al. Epileptic encephalopathy-causing mutations in DNM1 impair synaptic vesicle endocytosis. Neurol Genet 1, e4–e4 (2015).
- Ostrander, B. E. P. et al. Whole-genome analysis for effective clinical diagnosis and gene discovery in early infantile epileptic encephalopathy. npj Genomic Medicine 3, 22 (2018).
- Veeramah, K. R. et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 54, 1270–1281 (2013).
- Kearney, H., Byrne, S., Cavalleri, G. L. & Delanty, N. Tackling Epilepsy with High-definition Precision Medicine: A Review. JAMA Neurology 76 1109–1116 (2019).
- Guerrini, R., Balestrini, S., Wirrell, E. C. & Walker, M. C. Monogenic Epilepsies. Neurology 97, 817 (2021).
- Catterall, W. A., Kalume, F. & Oakley, J. C. NaV1.1 channels and epilepsy. J Physiol 588, 1849–1859 (2010).
- Richards, K. L. et al. Selective Na(V)1.1 activation rescues Dravet syndrome mice from seizures and premature death. Proc Natl Acad Sci U S A 115, E8077–E8085 (2018).
- Ridler, C. Cannabidiol reduces seizure frequency in Dravet syndrome. Nature Reviews Neurology 13, 383 (2017).
- Chaplin, S. Fenfluramine as add-on therapy for Dravet syndrome. Prescriber 32, 34–35 (2021).
- Guerrini, R. et al. Lamotrigine and Seizure Aggravation in Severe Myoclonic Epilepsy. Epilepsia 39, 508–512 (1998).
- Brunklaus, A., Ellis, R., Reavey, E., Forbes, G. H. & Zuberi, S. M. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 135, 2329–2336 (2012).
- Marini, C. et al. PRRT2 mutations in familial infantile seizures, paroxysmal dyskinesia, and hemiplegic migraine. Neurology 79, 2109–2114 (2012).
- Coleman, J. et al. PRRT2 Regulates Synaptic Fusion by Directly Modulating SNARE Complex Assembly. Cell Rep 22, 820–831 (2018).
- Fruscione, F. et al. PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain 141, 1000–1016 (2018).
- Numoto, S. et al. Sodium channel blockers are effective for benign infantile epilepsy. Seizure 92, 207–210 (2021).
- Ishida, S. et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet 45, 552–555 (2013).
- de Fusco, A. et al. Acute knockdown of Depdc5 leads to synaptic defects in mTOR-related epileptogenesis. Neurobiology of Disease 139, 104822 (2020).
- Klofas, L. K., Short, B. P., Zhou, C. & Carson, R. P. Prevention of premature death and seizures in a Depdc5 mouse epilepsy model through inhibition of mTORC1. Hum Mol Genet 29, 1365–1377 (2020).
- Yuskaitis, C. J. et al. Chronic mTORC1 inhibition rescues behavioral and biochemical deficits resulting from neuronal Depdc5 loss in mice. Hum Mol Genet 28, 2952–2964 (2019).
Publication History
Submitted: April 02, 2023
Accepted: April 20, 2023
Published: May 01, 2023
Identification
D-0115
Citation
Pradip Rijal (2023). Advances of NGS in Understanding of Epilepsy Genetics and Recent Discoveries of Gene in Monogenic Epilepsies. Dinkum Journal of Medical Innovations, 2(05):170-181.
Copyright
© 2023 DJMI. All rights reserved