

")
Publication History
Submitted: April 05, 2023
Accepted: April 20, 2023
Published: May 01, 2023
Identification
D-0118
Citation
Sarah Gul Rehman & Deepak Singh (2023). Thalassemia Consequences and Survival Rates in South Asian Kids and Adults. Dinkum Journal of Medical Innovations, 2(05):194-200.
Copyright
© 2023 DJMI. All rights reserved
194-200
Thalassemia Consequences and Survival Rates in South Asian Kids and AdultsReview Article
Sarah Gul Rehman 1, Deepak Singh 2*
- King Edward Medical University, Lahore, Pakistan; saragul@gmail.com
- Sunsari Technical College, Nepal; Deepaksingh34@gmail.com
* Correspondence: Deepaksingh34@gmail.com
Abstract: In order to assess the problems and life expectancy of patients with thalassemia (TDT, NTDT) in South Asia. A methodical approach was employed, adhering to PRISMA principles. Using specific keywords, the PubMed and Google Scholar databases were searched to find 8 papers. Included were studies discussing life expectancy in South Asian people and the difficulties of thalassemia. A methodical strategy was used to acquire and synthesise data in order to assess the effect of thalassemia on the health and life expectancy of patients. According to eight carefully chosen articles on complications in thalassemia patients (TDT and NTDT), encompassing both adult and paediatric age groups in South Asia, the following are the main complications that arise from iron overload in thalassemia major patients following multiple transfusions: hepatomegaly (41%), splenomegaly (14-56%), endocrine complications, hypothyroidism (4–25%), cardiac complications (10–12%) that rapidly shorten life expectancy, abnormalities of the bones, such as osteoporosis (59%), infections of the hep-C (3-67%) and hep-B, and extramedullary hematopoiesis. While transfusions are less common, NTDT/thalassemia intermedia also exhibits a wide range of clinical consequences. Iron overload, insufficient erythropoiesis, and hypercoagulability are NTDT/TI consequences. osteoporosis (81%), thrombosis, pulmonary hypertension, and extramedullary hematopoiesis (20%) are bone abnormalities. In South Asia, thalassemia major/intermedia has been linked to severe complications that shorten the disease’s prognosis. According to the study, thalassemia patients’ life expectancy is significantly increased by early diagnosis and prompt access to specialised care and treatment.
Keywords: life expectancy, thalassemia, survival rates, South Asia
- INTRODUCTION
Any inherited genetic abnormalities that affect the 𝛼-or 𝛽-globin chains’ ability to produce 𝛼-or 𝛽-globin chains, and consequently, appropriate erythropoiesis and hemoglobin’s ability to carry oxygen, are referred to as thalassaemia. Known as 𝛼- and 𝛽-thalassemia, this disease is inherited as an autosomal recessive trait. Patients with thalassemia disease often have ineffective erythropoiesis and prolonged hemolytic anaemia as a result of an imbalanced globin synthesis. (Source: ) Based on genetic and clinical features, 𝛽-thalassemia is divided into three types: thalassemia major, thalassemia intermedia, and thalassemia minor. Individuals with 𝛽-thalassemia major (TM) have two incorrect copies of the 𝛽-globin chain and, during the first two years of life, they suffer from severe transfusion-dependent microcytic anaemia. People who have thalassemia minor, often known as the carrier status, typically exhibit no clinical symptoms and have one defective copy of the 𝛽 -globin chain. [1-4] Beta thalassemia intermedia (𝛽-TI) is a disease of intermediate severity, with milder clinical symptoms and a delayed onset of microcytic anaemia when compared to 𝛽-TM. It is a subtype of 𝛼-thalassemia intermedia, or non-transfusion-dependent thalassemia (NTDT).[/2] Almost 80 percent of these births occur in developing nations. The most cautious estimates place the number of people with a major haemoglobin variant at least 5.2% of the global population, or 360 million [3]. Globally, there are approximately 100 million carriers of beta-thalassemia, with a 1.5% prevalence [4]. The world’s most populous regions are Africa, all Mediterranean nations, the Middle East, the Indian subcontinent, and Southeast Asia [3, 5]. Worldwide, more than 50,000 newborns are diagnosed with beta-thalassemia major and HbE beta-thalassemia, two severe forms of the disease. About 1.7 billion people, or 23% of the world’s population, live in South Asia, a hemoglobinopathies location [4,5], Within Pakistan, thalassemia is regarded as a serious public health issue due to its carrier prevalence of approximately 5%. Approximately five thousand new cases of beta-thalassemia major (TM) are detected each year [6]. Patients with thalassemia are currently divided into two groups based on the requirements for red blood cell transfusion: those with transfusion-dependent thalassemia (TDT) and those without (NTDT). Individuals with severe forms of thalassemia, such as homozygous β0-thalassemia or haemoglobin E /β-thalassemia, who suffer from transfusion-dependent thalassemia (TDT), depend on regular blood transfusions to survive. When undergoing specific medical procedures, such as pregnancy, surgery, or infection, patients with non-transfusion-dependent thalassemia need infrequent red blood cell transfusions. Patients with non-thalassemia disease therapy (NTDT) comprise individuals with moderate thalassemias, such as haemoglobin H sickness, and certain cases of haemoglobin E/β-thalassemia. #7, #8 Frequent blood transfusions increase the body’s iron stores and precipitate iron in vital organs such the heart, liver, and glands. This can cause serious issues such hypothyroidism, hyperparathyroidism, cirrhosis, diabetes, and cardiovascular diseases.In [9] The two primary kinds of thalassemia are thalassemia major and thalassemia intermedia. Both pose serious health risks to those who have them, especially in South Asia where thalassemia is very common. Even though thalassemia is very common in South Asia, little is known about the full range of complications related to thalassemia major and thalassemia intermedia, including how they affect the life expectancy of affected individuals in the region. In this region, there is a conspicuous dearth of thorough systematic reviews that specifically address the problems of thalassemia and life expectancy among children and adults. Research that is now available usually focuses on thalassemia in certain nations or globally. Thus, the purpose of this study was to assess the problems and life expectancy of thalassemia (TDT, NTDT) patients in South Asia.
- IMATERIALS AND METHODS
PubMed and Google Scholar were the two databases that were searched for this systematic review. Articles published between January 2014 and July 2023 were found through a search. MeSH and Non-mesh were among the keywords used. Southern Asia In addition to thalassemia, complications, iron overload, abnormalities of the bones, endocrine illnesses, cardiac complications, and life expectancy, boolean operators AND OR were used. Not included were articles that weren’t published in English. Furthermore, the method by which the studies were selected did not. Viewpoints, case reports, case series, meta-analyses, clinical trials (RCTS), and grey literature are all included. To find the articles, the pertinent databases were searched using the inclusion criteria listed below. The essay was published in the ten years prior. description of a geographic area. South Asian-related articles were among them. All age groups were encompassed. Case-control, cross-sectional, systematic, and scooping reviews were among them. After removing duplicates, the authors (M.A., N.B., and M.S.) confirmed the titles and abstracts in accordance with the qualifying requirements. The selected articles advanced to the following round following a comprehensive text assessment. The authors selected the pieces. In case there was any uncertainty, the other authors (S.A., A.A., and KJ) evaluated the papers to confirm their eligibility. Using the PRISMA principles, a Prisma flow sheet was made in order to extract the material. All relevant data was gathered by adhering to the selection criteria. The summary table includes the names, titles, and publication dates of the authors. In particular, details about TDT and NTDT issues were given. With the aid of the table structure, the authors were able to finish a comprehensive over-review of the data selected in the first step.
- RESULTS AND DISCUSSION
The authors’ process for choosing the papers is depicted in Figure 1’s PRISMA flow diagram. 940 articles were found after the authors conducted searches in two databases, PubMed and Google Scholar. The authors eliminated ninety duplicates from these first records. There were still 850 items to be reviewed. We reduced the total number of articles to 45 after screening titles and abstracts by following exclusion criteria. articles for which the complete text is not accessible. Excluded from consideration were RTCs, clinical trials, book chapters, and articles published in languages other than English. After giving the remaining articles a thorough reading, the final eight publications that satisfied the inclusion criteria—which included research referencing thalassemia problems and a specific geographic area—were chosen. Articles about South Asia were among them. The following are the main complications that arise in patients with thalassemia major due to iron overload after multiple transfusions: hepatomegaly (41%), splenomegaly (14-56%), endocrine complications (such as short stature, 25%), hypothyroidism (4–25%), cardiac complications (10–12%) that shorten life expectancy quickly, bone abnormalities (such as osteoporosis, 59%), infections (Hep-C, 3-67%), and extramedullary hematopoiesis. These findings are based on eight carefully chosen articles that include complications in thalassemia patients (TDT and NTDT) across all age groups, children, and adults in South Asia. Although thalassemia intermedia and NTDT both exhibit Even with fewer transfusions, there were a number of clinical issues.
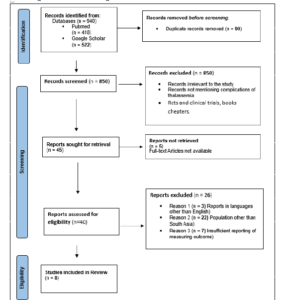
Figure 1: PRISMA Flow chart
According to the study’s findings, iron overload and hemolysis from increased intestinal absorption cause complications in thalassemia major patients (TDT and NTDT) of all ages in South Asia. These complications are caused by repeated blood transfusions. In individuals with thalassemia, iron overload and elevated cardiac output primarily compromise cardiac structure and function. This leads to a range of complications, including splenomegaly, hepatomegaly, endocrine disorders, hypothyroidism, and heart problems that drastically reduce life expectancy. Additionally, abnormalities in bone structure like osteoporosis, blood transfusion infections like Hep-C and Hep-B, and extramedullary hemopoiesis are also common. However, even though NTDT/thalassemia intermedia receive fewer transfusions, it nonetheless presents with serious clinical issues. Complications in NTDT/TI are brought on by a confluence of many pathophysiological factors, including iron overload, hypercoagulability, and inadequate erythropoiesis. Osteoporosis, extramedullary hemopoiesis, thrombosis, pulmonary hypertension, and abnormalities of the bones are among the main consequences of thalassemia intermedia. A study conducted in Iran found that individuals with beta-thalassemia who had elevated blood ferritin levels had a two to four times higher risk of developing hepatic or cardiac iron overload, which can result in considerable mortality [14]. Similarly, a Turkish study discovered that individuals with beta-thalassemia who had increased blood ferritin levels needed to be regularly monitored for different endocrinopathies [15]. The life expectancy of patients with beta-thalasemia has been greatly increased by safer transfusion practises and adjuvant chelation therapy, despite the poor disease development of the condition [16, 17]. Extended periods of living with complications can be excruciating for the patient and expensive for the healthcare system as a whole[18]. The majority of large thalassemic patients will live long lives, although the likelihood of dying soon decreases when heart failure symptoms appear [19]. For people with thalassemia, heart failure is still considered to be one of the main reasons of death. Early identification of heart dysfunction is therefore essential. Twenty [20] The offspring of a heterozygous marriage are at 25% risk of having children with -thalassemia major, 25% risk of heterozygous children, and 50% chance of normal children [21]. A study conducted in Pakistan revealed that in nine out of fourteen thalassemia patients following splenectomy, the issue was wound and respiratory infections.When children with significant splenomegaly receive the right perioperative care, splenectomy is a safe and effective surgical procedure [23]. Children with thalassemia experience serious psychosocial issues. Sexual problems and psychosis are common in teenagers [24]. Anaemia is a serious issue for expectant mothers and has been linked to unfavourable pregnancy outcomes. In Punjab, Pakistan, a significant problem that has been identified is the absence of established protocols for case management and laboratory identification of thalassemia patients, as well as a lack of inter- and intra-institutional coordination and referral linkage. A new study has shown that, contrary to earlier assumptions, 𝛽-TI is not a minor illness, but rather one associated with increased morbidity and a wider range of organ dysfunction and repercussions. Due to the high frequency of endocrine and bone issues associated with this illness, ongoing treatment, monitoring, and follow-up are necessary. Early detection of these consequences, the application of suitable treatment, such as chelation therapy and transfusion regimens, and the tailored management of each complication are essential to the effectiveness of care. This study examined the problems and life expectancy of patients with thalassemia major and thalassemia intermedia in South Asia. A thorough search technique was used in the systematic review to locate pertinent studies across several databases, including Pubmed and Google Scholar. The inclusion and exclusion criteria for the relevant research were clearly defined in this analysis. Pie charts and tables were used in the study’s descriptive summary to convey the findings in an organised and understandable manner. This guarantees that readers will be able to comprehend the results and their significance with ease. The systematic review’s included papers show significant variation in terms of research design, patient characteristics, therapies, and outcomes evaluated, which is one of its limitations. This heterogeneity may limit the findings’ comparability and generalizability and may hinder the capacity to do a meta-analysis. Publication bias is a potential, in which research with statistically significant or positive outcomes are more likely to be published, but non-significant or negative results may be underrepresented. We do not measure bias in this study. The review might only include research that have been written in a particular language, like English. One risk is that pertinent studies written in other languages would go unnoticed, which could result in a representation of the literature that is only partially complete. A number of the papers that we have included in our review article have not provided all necessary information for analysis or all pertinent data. This may affect the review’s comprehensiveness or restrict the capacity to pull data for particular outcomes. The evaluation may predominantly concentrate on research conducted in certain South Asian nations because of data accessibility issues and linguistic constraints. This can lead to an unequal distribution of thalassemia cases throughout the area. Some thalassemia studies have not recorded all pertinent results or have only examined specific facets of the disease, omitting important consequences or contributing factors. Comparative studies comparing various South Asian nations and areas are advised as a means of investigating differences in the problems and life expectancy associated with thalassemia. It is important to plan and carry out interventional studies to assess how well various treatment modalities manage side effects and extend the life expectancy of thalassemia patients. These research may contribute to the development of evidence-based therapeutic guidelines and the identification of the best treatment plans. The advantages and financial viability of early thalassemia diagnosis and screening initiatives in South Asia warrant further investigation. to underline how crucial carrier screening and genetic counselling are to enabling educated family planning decisions and lowering the incidence of thalassemia. Thus, there may be obstacles in the way of receiving specialised care and treatment for thalassemia in many parts of South Asia. The usefulness of health education and awareness campaigns aimed at the general public, healthcare professionals, and policymakers to increase knowledge about thalassemia, its complications, and preventive measures should be assessed. The impact of multidisciplinary care teams, including haematologists, paediatricians, psychologists, and social workers, on improving outcomes for thalassemia patients should also be evaluated. More research should be done to determine the variables that affect thalassemia patients’ life expectancy, such as socioeconomic position, access to healthcare, management of their condition, and adherence to their treatment plans. Knowing these variables can help develop better results and longer life expectancy techniques.
- CONCLUSION
The review highlighted the significant burden of Thalassemia Major and Thalassemia Intermedia complications in South Asia. Iron overload, organ damage, blood transfusion infections, and cardiac, endocrine, and bone problems were recognized as key concerns, emphasizing the importance of comprehensive therapy. Thalassemia has been found to have a significant impact on the life expectancy of affected people in South Asia. The analysis found that early diagnosis and timely access to specialized care and treatment play an important role in enhancing thalassemia patients’ life expectancy. The findings of the study have significant public health implications. Early diagnosis, genetic counseling, and greater access to specialized care have emerged as essential elements of effective thalassemia control and preventive measures.
REFERENCES
- Taher AT, Radwan A, Viprakasit V. When to consider transfusion therapy for patients with non‐transfusion‐dependent thalassaemia. Vox sangu-inis. 2015;108(1):1-10.
- Inati A, Noureldine MA, Mansour A, Abbas HA. Endocrine and bone complications in β-thalas-semia intermedia: current understanding and treatment. BioMed researchs international. 2015;-015(1):1-10.
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86-(1):480–7.
- Colah R, Gorakshakar A, Nadkarni A. Global burden, distribution and prevention of β-thalas-semias and hemoglobin E disorders. Expert Rev Hematol. 2010;3(1):103–17.
- Hossain MS, Raheem E, Sultana TA, Ferdous S, Nahar N, Islam S, et al. Thalassemias in South Asia: clinical lessons learnt from Bangladesh. Orphanet journal of rare diseases. 2017;12(1):1-9.
- Yasmeen H, Hasnain S. Epidemiology and risk factors of transfusion transmitted infections in thalassemia major: a multicenter study in Pakistan. Hematology, transfusion and cell therapy. 2019;41(1): 316-23.
- Chuncharunee S, Teawtrakul N, Siritanaratkul N, Chueamuangphan N. Review of disease-related complications and management in adult patients with thalassemia: A multi-center study in Thailand. PLoS One. 2019;14(3):0214148.
- Mahmoodi Z. Prevalence of cardiac complications in patients with major thalassemia in iranian patients: a systematic review and meta-analysis. IJPBA. 2019;7(6):59-63.
- Abu-Shaheen A, Heena H, Nofal A, Abdelmoety DA, Almatary A, Alsheef M, et al. Epidemiology of thalassemia in Gulf Cooperation Council countries: a systematic review. BioMed research international. 2020;2020(1):1-15.
- Maryam Majeed, Rubab Ali, Umm-e-Aimen & Aqsa Jabeen. Medical Staff, Workplace Bullying and Its Effects on the Performance. Dinkum Journal of Medical Innovations, 2(04):120-125.
- Marie Diack. Factors Influencing Relational Violence in Nepali Married Relationships. Dinkum Journal of Medical Innovations, 2(04):126-133.
- Gul Rukh Malik, Rubab Zahra & Tahir Rana. Autism Spectrum Disorders Frequency in Asian Countries. Dinkum Journal of Medical Innovations, 2(04):134-139.
- Parshu Ram Chaudhary, Asbin Bandhari & Parshu Kirby. Aeroallergens and Significant Environmental Pollutants: Aeroallergen Sensitivity Symptoms. Dinkum Journal of Medical Innovations, 2(04):140-149.
- Imran Mehfooz Khan, Aron Kumar & Pradeep Sahejpal. Extensive Management Medical Incidents Provided to Gallbladder Cancer Using Cholecystectomy Specimen. Dinkum Journal of Medical Innovations, 2(04):150-156.
- Ahmed S, Saleem M, Modell B, Petrou M. Screening extended families for genetic hemo-globin disorders in Pakistan. N Engl J Med. 2002;347(1): 1162-8.
- Mahmoodi Z. The association of stress, HTN and DM during pregnancy. Int. J. Curr. Res. Med. Sci. 2017;3(12):63-6.
- Chahkandi T, Kazemi T, Jalili A, Ghaderi F. Evaluation of cardiac function in patients with major beta thalassemia in Birjand. Scientific Journal of Iranian Blood Transfusion Organization. 2013 ;10(1):86-92.
- Hajipour M, Mehrabi Y, Mortezaeian H, Mohseni P, Aghighi M, Hantooshzadeh R, et al. Risk Factors Associated With Heart Failure in Patients With β-Thalassemia. Iranian Heart Journal. 2022;23(3):88-96.
- Anjum S, Tayyab M, Hussain Z, Shah H. Detection of β-Thalassaemia Trait: A Study of Fifty Families. Annals of King Edward Medical University. 2000;6(3):275-7
- Majid A, Zahra F, Waheed M, Manan J. Exper-ience of splenectomy in Thalassemia. Annals of King Edward Medical University.2004;10-(1):66-7.
- Zahra F, Ashraf M, Aslam M, Deen Q, Mannan J. Complications associated with splenectomy in thalassemia. Annals of King Edward Medical University. 2005;11(4):507-9.
- Saqlain S, Arif S, Batool S, Rehman S, Usman M, Sarfraz MU, et al. Psychosocial Problems Faced by Thalassemia Patients and their Parents. Journal of Society of Prevention, Advocacy and Research KEMU. 2022;1(3):1-10.
- Sajid A, Sajid A. Frequency of Anemia and Obstetric Outcome in Young Primigravida. Annals of King Edward Medical University. 2017;23-(4):531-5
- Majrooh MA, Fatima N. Status of Thalassemia and Haemophilia Services in Existing Health Care Delivery System of the Punjab. Annals of King Edward Medical University. 2002;8(1):39-42.
Publication History
Submitted: April 05, 2023
Accepted: April 20, 2023
Published: May 01, 2023
Identification
D-0118
Citation
Sarah Gul Rehman & Deepak Singh (2023). Thalassemia Consequences and Survival Rates in South Asian Kids and Adults. Dinkum Journal of Medical Innovations, 2(05):194-200.
Copyright
© 2023 DJMI. All rights reserved